






Qu'est-ce que le Rhabdomyosarcome Embryonnaire et Comment est-il Diagnostiqué?

-
Comprendre le Rhabdomyosarcome Embryonnaire
-
Épidémiologie et Données Statistiques
-
Caractéristiques du Rhabdomyosarcome Embryonnaire
-
La Variante Botryoïde (Sarcoma Botryoides)
-
Facteurs de Risque et Prédispositions
-
Manifestations Cliniques et Symptômes
-
Méthodes de Diagnostic du Rhabdomyosarcome Embryonnaire
-
Stadification et Évaluation du Pronostic
-
Approches Thérapeutiques Actuelles
-
Suivi à Long Terme et Qualité de Vie
-
Conclusion

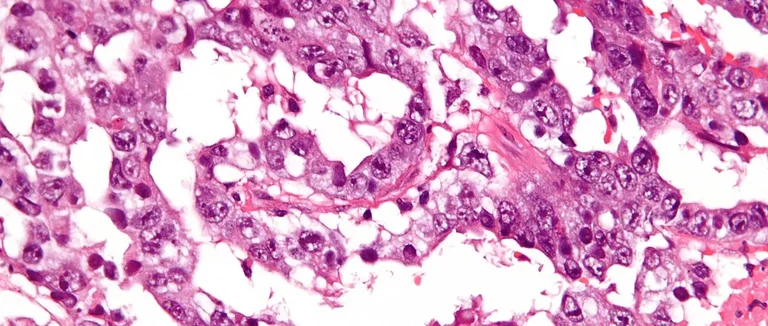
Le rhabdomyosarcome embryonnaire (ERMS) est un cancer rare chez les enfants. Il vient des cellules musculaires embryonnaires. La tumeur peut apparaître de différentes manières selon son emplacement.
Les symptômes peuvent varier. Ils peuvent inclure des douleurs, des gonflements ou des difficultés à bouger. Un diagnostic précis est essentiel pour trouver le meilleur traitement.
La Fondation St. Baldrick et l'American Cancer Society offrent des infos utiles sur cette maladie. Un diagnostic et un traitement tôt sont cruciaux pour améliorer les chances de guérison. Nous offrons des soins personnalisés pour les enfants touchés par cette maladie rare.
Points Clés à Retenir
● Définition et incidence du rhabdomyosarcome embryonnaire
● Symptômes et diagnostic de l'ERMS
● Importance d'un traitement précoce
● Soins multidisciplinaires pour les enfants atteints
● Ressources et soutien pour les familles
Comprendre le Rhabdomyosarcome Embryonnaire
Le rhabdomyosarcome embryonnaire vient d'anomalies dans les cellules mésenchymateuses primitives. C'est un cancer qui affecte les enfants. Il se développe à partir de cellules qui devraient devenir des muscles squelettiques.
Origine cellulaire et développement
Les cellules mésenchymateuses primitives ne se transforment pas correctement en muscles. Elles prolifèrent de façon incontrôlée, créant des tumeurs malignes.
Comprendre l'origine cellulaire aide à trouver des traitements ciblés. Des études montrent que des mutations génétiques jouent un rôle dans l'apparition de l'ERMS.
Place dans la classification des sarcomes pédiatriques
Le rhabdomyosarcome embryonnaire est le plus fréquent chez les enfants. Il représente 60-70% des cas chez les moins de 10 ans.
| Type de Sarcome | Fréquence chez les Enfants | Caractéristiques |
| Rhabdomyosarcome Embryonnaire | 60-70% | Développement à partir de cellules embryonnaires |
| Rhabdomyosarcome Alvéolaire | 20-30% | Caractérisé par des structures alvéolaires |
| Autres Types | <10% | Comprend divers sous-types rares |
La classification des sarcomes pédiatriques est cruciale. Elle aide à déterminer le pronostic et le traitement. Le rhabdomyosarcome embryonnaire a un meilleur pronostic, surtout si diagnostiqué tôt.
Épidémiologie et Données Statistiques
Les données sur le rhabdomyosarcome embryonnaire sont très utiles. Elles nous aident à comprendre sa fréquence et qui est touché. Nous allons voir les chiffres pour saisir mieux l'ERMS.
Incidence aux États-Unis et dans le monde
Le rhabdomyosarcome embryonnaire est le type le plus fréquent, représentant 55-57% des cas. Aux États-Unis, environ 350 enfants sont diagnostiqués chaque année. Le taux d'incidence varie dans le monde, selon les populations et les méthodes de détection.
Distribution démographique et facteurs d'âge
L'ERMS touche surtout les enfants, surtout les garçons. Il est le plus fréquent chez les enfants de moins de 10 ans. Les âges influencent beaucoup l'épidémiologie de l'ERMS.
Les facteurs génétiques et les conditions environnementales ont un impact sur l'incidence de l'ERMS. Les recherches épidémiologiques sont cruciales pour mieux combattre cette maladie.
Caractéristiques du Rhabdomyosarcome Embryonnaire
Le rhabdomyosarcome embryonnaire (ERMS) est un cancer rare. Il a des caractéristiques uniques. Ces traits sont essentiels pour diagnostiquer et classer ce sarcome.
Propriétés histologiques distinctives
L'ERMS se distingue par des cellules primitives. Elles montrent une différenciation vers les muscles squelettiques. Les cellules peuvent être rondes, ovales ou allongées avec des noyaux grands.
Les cellules rhabdomyoblastiques sont clés pour l'ERMS. Elles ont des caractéristiques musculaires. Elles contiennent des filaments et des stries Z, montrant leur différenciation musculaire.
Comparaison avec d'autres sous-types de rhabdomyosarcome
L'ERMS est un sous-type de rhabdomyosarcome. Il y a aussi le rhabdomyosarcome alvéolaire et le rhabdomyosarcome à cellules fusiformes. L'ERMS se distingue par sa morphologie variée et sa différenciation musculaire forte.
Le rhabdomyosarcome alvéolaire a une architecture alvéolaire. Il a des cellules avec des noyaux ronds et peu de différenciation. La distinction entre ces sous-types est cruciale pour le pronostic et le traitement.
La Variante Botryoïde (Sarcoma Botryoides)
La variante botryoïde est un type de rhabdomyosarcome embryonnaire. Elle se distingue par son apparence et ses sites d'apparition. Cette tumeur maligne se développe souvent sous les épithéliums muqueux des organes creux.
Caractéristiques spécifiques et sites d'apparition
Elle se caractérise par sa croissance en masses polypoïdes ou grappes de raisin. Cela lui a valu le nom de "sarcome botryoïde". Elle se développe surtout dans les organes creux comme la vessie et le vagin.
Les caractéristiques cliniques de la variante botryoïde comprennent :
● Apparition dans les organes creux
● Croissance sous forme de masses polypoïdes
● Fréquence chez les enfants et les adolescents
Présentation clinique particulière
La présentation clinique de la variante botryoïde dépend de sa localisation. Par exemple, dans la vessie, elle peut causer des symptômes urinaires. Dans le vagin, elle peut provoquer des saignements vaginaux anormaux.
| Localisation | Symptômes Cliniques |
| Vessie | Hématurie, dysurie |
| Vagin | Saignements vaginaux anormaux |
| Voies biliaires | Ictère, douleurs abdominales |
Reconnaître ces caractéristiques cliniques est crucial pour un diagnostic précoce. Cela permet une prise en charge adéquate de la variante botryoïde du rhabdomyosarcome embryonnaire.
Facteurs de Risque et Prédispositions
Les recherches sur les facteurs de risque et les prédispositions ouvrent de nouvelles perspectives dans la prévention et le traitement de l'ERMS. Nous examinons ici les éléments qui augmentent la susceptibilité à ce type de cancer.
Facteurs génétiques associés
Les facteurs génétiques jouent un rôle crucial dans le développement de l'ERMS. Certaines mutations génétiques peuvent prédisposer les individus à ce cancer. Par exemple, le syndrome de Li-Fraumeni, causé par des mutations dans le gène TP53, est connu pour augmenter le risque de développer divers cancers, y compris l'ERMS.
Des études ont montré que les personnes ayant des antécédents familiaux de cancer peuvent avoir un risque accru de développer l'ERMS. Il est donc essentiel de comprendre ces facteurs génétiques pour une détection précoce et une prise en charge appropriée.
Syndromes héréditaires et conditions prédisposantes
Outre les facteurs génétiques directs, certains syndromes héréditaires et conditions médicales peuvent prédisposer les individus à l'ERMS. Le syndrome de Li-Fraumeni, comme mentionné, est l'un de ces syndromes. D'autres conditions, telles que la neurofibromatose de type 1, peuvent également augmenter le risque.
Il est crucial de reconnaître ces syndromes et conditions pour offrir une surveillance et des soins appropriés. Le tableau suivant résume quelques-uns des syndromes héréditaires et conditions prédisposantes associés à l'ERMS.
| Syndrome ou Condition | Description | Risque Associé |
| Syndrome de Li-Fraumeni | Mutation du gène TP53 | Risque accru de divers cancers, y compris l'ERMS |
| Neurofibromatose de type 1 | Mutation du gène NF1 | Risque accru de tumeurs du système nerveux et potentiellement de l'ERMS |
| Syndrome de Beckwith-Wiedemann | Anomalie génétique sur le chromosome 11 | Risque accru de tumeurs embryonnaires, y compris potentiellement l'ERMS |
En comprenant ces facteurs de risque et prédispositions, nous pouvons améliorer la détection précoce et la prise en charge de l'ERMS. Les recherches continuent d'évoluer, offrant de nouvelles perspectives sur la prévention et le traitement de ce cancer complexe.
Manifestations Cliniques et Symptômes
La maladie ERMS se manifeste de différentes manières selon l'endroit où la tumeur se trouve. Les symptômes peuvent être des saignements de nez, des problèmes urinaires ou des douleurs abdominales.
Signes précoces selon la localisation anatomique
Les premiers signes de l'ERMS changent selon l'endroit de la tumeur. Par exemple, les tumeurs dans la tête et le cou peuvent causer des douleurs faciales ou des problèmes à avaler. Les tumeurs dans l'abdomen ou le bassin peuvent donner des douleurs, des nausées ou des problèmes urinaires.
Voici un tableau des signes précoces de l'ERMS selon l'endroit de la tumeur :
| Localisation Anatomique | Signes Précoces |
| Tête et Cou | Douleurs faciales, difficultés à avaler, changements de voix |
| Abdomen/Pelvis | Douleurs abdominales, nausées, difficultés urinaires |
| Membres | Gonflement, douleurs, limitation de la mobilité |
Progression de la maladie et complications
La maladie ERMS peut se propager et causer des complications sévères. Ces complications peuvent inclure des problèmes respiratoires, des troubles neurologiques ou des défaillances d'organes. Cela dépend de l'endroit et de l'étendue de la maladie.
Il est essentiel de diagnostiquer et de traiter l'ERMS rapidement. Cela aide à réduire les risques de complications et à améliorer les chances de guérison.
Méthodes de Diagnostic du Rhabdomyosarcome Embryonnaire
Diagnostiquer le rhabdomyosarcome embryonnaire demande une approche à plusieurs niveaux. Nous employons des méthodes cliniques et paracliniques pour obtenir une précision maximale.
Évaluation clinique initiale
La première étape est d'identifier les signes et symptômes de l'ERMS. Un examen physique minutieux est crucial. Il permet d'évaluer la taille et la localisation de la tumeur, ainsi que de rechercher des métastases.
Techniques d'imagerie médicale (IRM, TDM, TEP)
Les techniques d'imagerie médicale sont essentielles pour le diagnostic. L'IRM est particulièrement utile pour voir l'étendue de la tumeur et sa relation avec les tissus alentours.
La TDM aide à détecter les métastases pulmonaires et les calcifications tumorales. La TEP permet d'évaluer l'activité métabolique de la tumeur et de repérer les métastases à distance.
Biopsie et analyse histopathologique
La biopsie est cruciale pour confirmer le diagnostic. Nous prenons des échantillons tissulaires pour les analyser au microscope. Cela confirme la présence de cellules tumorales.
L'analyse histopathologique classe la tumeur selon son type et son grade. Les caractéristiques de l'ERMS incluent des cellules avec des noyaux hyperchromatiques et un cytoplasme éosinophile.
| Caractéristiques | ERMS | Autres Sarcomes |
| Cellules tumorales | Petites, rondes, avec noyaux hyperchromatiques | Variables selon le type de sarcome |
| Marqueurs immunohistochimiques | Desmin, MyoD1, myogénine positifs | Variables selon le type de sarcome |
Marqueurs immunohistochimiques et tests moléculaires
Les marqueurs immunohistochimiques sont clés pour confirmer l'ERMS. Desmin, MyoD1 et myogénine sont souvent présents dans les cellules tumorales.
Les tests moléculaires identifient des anomalies génétiques spécifiques de l'ERMS, comme la translocation PAX-FOXO1.
En combinant ces méthodes, nous obtenons un diagnostic précis. Cela nous permet de planifier un traitement adapté pour les patients.
Stadification et Évaluation du Pronostic
La stadification et l'évaluation du pronostic sont essentielles dans la gestion de l'ERMS. Elles permettent de connaître la gravité de la maladie. Elles aident aussi à choisir le meilleur traitement.
Systèmes de classification clinique
Les systèmes de classification clinique évaluent l'étendue de la maladie. Ils déterminent aussi le pronostic. Le système TNM est le plus utilisé pour l'ERMS. Il considère la taille et l'emplacement de la tumeur, ainsi que l'atteinte des ganglions lymphatiques et la présence de métastases.
La classification clinique de l'Intergroup Rhabdomyosarcoma Study (IRS) est aussi importante. Elle catégorise les patients selon l'étendue de la maladie et la réponse au traitement. Cela aide à identifier les patients à haut risque qui nécessitent des traitements plus intensifs.
Groupes de risque et implications thérapeutiques
Les patients d'ERMS sont classés en groupes de risque. Ces groupes sont déterminés par l'âge, la localisation et la taille de la tumeur, ainsi que la présence de métastases. Ces classifications guident les décisions thérapeutiques et aident à prédire le pronostic.
Par exemple, les patients avec une tumeur localisée et sans métastase ont un meilleur pronostic. Les traitements sont donc adaptés en fonction du groupe de risque. Les patients à haut risque reçoivent des traitements plus intensifs.
Facteurs influençant le pronostic
Plusieurs facteurs influencent le pronostic de l'ERMS. L'âge du patient, la localisation et la taille de la tumeur, ainsi que la réponse au traitement initial sont importants. Les patients diagnostiqués à un jeune âge ont souvent un meilleur pronostic.
La présence de certaines caractéristiques génétiques ou moléculaires peut aussi influencer le pronostic. Les avancées dans la biologie de l'ERMS aident à identifier de nouveaux facteurs pronostiques. Elles permettent de développer des stratégies de traitement ciblées.
Approches Thérapeutiques Actuelles
Les stratégies actuelles pour l'ERMS combinent plusieurs méthodes. Elles visent à améliorer les résultats pour les patients.
Stratégies chirurgicales
La chirurgie est souvent le premier traitement pour l'ERMS. Elle vise à enlever la tumeur primitive.
Les stratégies chirurgicales dépendent de la tumeur. Cela inclut sa localisation et sa taille.
● Résection large pour les tumeurs accessibles
● Chirurgie conservatrice pour préserver la fonction des organes
Protocoles de radiothérapie
La radiothérapie traite les résidus tumoraux après chirurgie. Elle est aussi utilisée pour les tumeurs inopérables.
Les protocoles de radiothérapie sont adaptés à chaque patient. Ils tiennent compte de l'âge et de la localisation de la tumeur.
| Âge du patient | Dose de radiothérapie |
| Moins de 5 ans | 30 Gy |
| 5 ans et plus | 40-50 Gy |
Régimes de chimiothérapie
La chimiothérapie est cruciale dans le traitement de l'ERMS. Elle réduit la taille de la tumeur avant la chirurgie ou traite les métastases.
Les régimes de chimiothérapie combinent plusieurs médicaments. Cela inclut la Vincristine, l'Actinomycine D et le Cyclophosphamide.
● Vincristine
● Actinomycine D
● Cyclophosphamide
Thérapies ciblées émergentes
Les thérapies ciblées sont une nouvelle approche pour l'ERMS. Elles visent spécifiquement les anomalies moléculaires.
Ces thérapies promettent d'améliorer les résultats. Elles visent aussi à réduire les effets secondaires.
Suivi à Long Terme et Qualité de Vie
Les patients qui ont survécu à l'ERMS ont besoin d'une surveillance continue. Cela affecte leur qualité de vie. Un suivi à long terme aide à détecter les récidives et à gérer les effets tardifs.
Surveillance des récidives
Surveiller les récidives est crucial. Les patients doivent être examinés régulièrement. Cela permet de traiter rapidement en cas de récidive.
Un plan de suivi personnalisé est recommandé. Il tient compte des risques et de la maladie. Les visites médicales, les analyses de sang, et les examens d'imagerie sont essentiels.
Gestion des effets tardifs du traitement
Les traitements contre l'ERMS peuvent avoir des effets tardifs. Ces effets peuvent affecter la santé et la qualité de vie. Il est important de les gérer.
| Effets Tardifs | Stratégies de Gestion |
| Problèmes cardiaques | Surveillance cardiaque régulière, modification du style de vie |
| Difficultés de croissance | Suivi de la croissance, interventions orthopédiques si nécessaire |
| Problèmes de fertilité | Conseil en fertilité, préservation de la fertilité |
Soutien psychosocial
Le soutien psychosocial est essentiel pour les survivants d'ERMS. Ils peuvent rencontrer des défis émotionnels et psychologiques. Nous offrons des conseils et un soutien pour les aider.
Le soutien peut inclure la thérapie individuelle ou de groupe. Il peut aussi inclure des ateliers sur le stress et l'adaptation. Notre but est d'améliorer leur qualité de vie.
Conclusion
L'ERMS est un cancer rare mais très grave. Il faut une approche combinée pour le traiter. Nous avons parlé de son origine, comment il est diagnostiqué, et les traitements disponibles.
Il est crucial de le détecter tôt et de le traiter correctement. Les avancées dans la chirurgie, la radiothérapie et la chimiothérapie offrent de nouvelles espoirs. Ces progrès sont importants pour les patients.
En résumé, l'ERMS demande beaucoup d'attention. Il est complexe et affecte beaucoup les patients, surtout les enfants. Il est essentiel que les professionnels travaillent ensemble pour offrir les meilleurs soins.
Foire Aux Questions
Le rhabdomyosarcome embryonnaire est un cancer rare chez les enfants. Il touche les tissus mous. C'est un type de rhabdomyosarcome, un cancer des muscles squelettiques.
Les symptômes de l'ERMS dépendent de l'endroit où la tumeur se trouve. On peut voir une masse ou un gonflement. Il y a aussi des douleurs, des problèmes à avaler ou à respirer.
Pour diagnostiquer l'ERMS, on fait d'abord une évaluation clinique. On utilise l'IRM et la TDM pour voir l'image. Une biopsie est analysée pour confirmer le diagnostic. Les marqueurs immunohistochimiques aident aussi.
Certains facteurs augmentent le risque d'ERMS. Des facteurs génétiques et des syndromes héréditaires jouent un rôle. Le syndrome de Li-Fraumeni, par exemple, augmente le risque.
Le traitement de l'ERMS varie selon le cas. On peut faire de la chirurgie, de la radiothérapie, de la chimiothérapie. Des thérapies ciblées émergentes sont aussi possibles. Le choix dépend de plusieurs facteurs.
Suivre les patients à long terme est essentiel. Cela aide à surveiller les récidives et à gérer les effets tardifs. Cela améliore aussi la qualité de vie des survivants.
La variante botryoïde est un type d'ERMS. Elle se trouve souvent dans les organes creux comme la vessie ou le vagin. Elle a des caractéristiques uniques.
Le pronostic de l'ERMS dépend de plusieurs facteurs. On regarde la stadification, les groupes de risque et l'âge du patient. Les systèmes de classification clinique aident à évaluer le pronostic.
* Le contenu de notre site web est uniquement destiné à des fins d'information. Veuillez consulter votre médecin pour obtenir un diagnostic et un traitement. Le contenu de la page ne contient pas d'informations sur les services de santé thérapeutiques de l'hôpital Liv.



